Hello fPocket
Show how to use fPocket to find locations to dock the ligands
fPocket - finds candidate locations to dock the ligand
Step 1: Get back into your lab environment.
Using the terminal, change to the directory with your target protein and activate your biolab conda environment. You may need to change the cd command below depending on your folder structure.
cd Documents/MolecularDocking/locks/
conda activate biolab
Step 2: Run fPocket on the same protein from the previous lesson, 1H9Z.
fpocket -f 1H9Z.pdb
The output text is underwhelming, the magic is in the files fPocket creates.
***** POCKET HUNTING BEGINS *****
***** POCKET HUNTING ENDS *****
fPocket has now created a folder called 1H9Z_out. This folder contains the following:
1H9Z_info.txt - Lists every pocket fPocket found, along with the stats for each one.
1H9Z_out.pdb - The original protein file, annotated with the pocket information so PyMOL can color the pockets.
1H9Z_pockets.pqr - The alpha spheres: the virtual marbles fPocket uses to fill each cavity.
1H9Z_PYMOL.sh - A script that, when run, opens PyMOL and loads the protein and ligand with some preset styling.
pockets - A folder containing detailed information about every pocket it found.
Step 3: Evaluating the output
Open 1H9Z_info.txt in a text editor to see its contents. Below is what fPocket found for the first pocket. fPocket ranks its best candidates at the top, so Pocket 1 is considered a stronger candidate than Pocket 40.
Pocket 1 :
Score : 2.370
Druggability Score : 0.996
Number of Alpha Spheres : 617
Total SASA : 714.062
Polar SASA : 224.280
Apolar SASA : 489.782
Volume : 4476.194
Mean local hydrophobic density : 67.850
Mean alpha sphere radius : 3.949
Mean alp. sph. solvent access : 0.467
Apolar alpha sphere proportion : 0.627
Hydrophobicity score: 35.510
Volume score: 4.388
Polarity score: 43
Charge score : 11
Proportion of polar atoms: 32.698
Alpha sphere density : 16.598
Cent. of mass - Alpha Sphere max dist: 40.507
Flexibility : 0.12
These are the most important stats:
Score - Overall score, higher is usually better
Druggability Score - Scale is 0 - 1. Higher is better, .5 and above is considered "druggable".
Volume - Volume of the cavity/pocket. It must be big enough so the ligand can fit into it.
Hydrophobicity - Higher numbers are often better, because of the hydrophobic effect.
Step 4: Put the data generated by fPocket to use.
We will take the top candidate, Pocket 1, and find its center. We will then try to dock the ligand to see
how well of a bond it makes, then view the results with PyMOL.
Switch to the pocket folder and open PyMOL:
cd ~/Documents/MolecularDocking/locks/1H9Z_out/pockets
pymol
Enter these commands into PyMOL's terminal
load pocket1_vert.pqr
pseudoatom pocket_center, pocket1_vert
print cmd.get_coords('pocket_center')
You will get some output that looks like this:
PyMOL>pseudoatom pocket_center, pocket1_vert
ObjMol: created pocket_center/PSDO/P/PSD`1 /PS1
PyMOL>print cmd.get_coords('pocket_center')
[[23.093771 8.962 14.027454]]
Update your config file with the new x, y, z coordinates. My config file is located here:
~/Documents/MolecularDocking/config.txt
receptor = ./locks/receptor.pdbqt
ligand = ./keys/warfarin.pdbqt
center_x = 23.09
center_y = 8.96
center_z = 14.03
size_x = 25.0
size_y = 25.0
size_z = 25.0
thread = 8000
search_depth = 10
energy_range = 4
Step 5: Run AutoDock-Vina-GPU-2
~/software/Vina-GPU-2.1/AutoDock-Vina-GPU-2.1/AutoDock-Vina-GPU-2-1 --config config.txt
Here is the output from AutoDock
#################################################################
# If you used AutoDockVina-GPU 2.1 in your work, please cite: #
# #
# Ding, Ji, et al. Vina-GPU 2.0: Further Accelerating AutoDock #
# Vina and Its Derivatives with Graphics Processing Units. #
# Journal of Chemical Information and Modeling (2023). #
# #
# DOI https://doi.org/10.1021/acs.jcim.2c01504 #
# #
# Shidi, Tang, Chen Ruiqi, Lin Mengru, Lin Qingde, #
# Zhu Yanxiang, Wu Jiansheng, Hu Haifeng, and Ling Ming. #
# Accelerating AutoDock Vina with GPUs. #
# Molecules 27.9 (2022): 3041. #
# #
# DOI https://doi.org/10.3390/molecules27093041 #
# #
# And also the origin AutoDock Vina paper: #
# O. Trott, A. J. Olson, #
# AutoDock Vina: improving the speed and accuracy of docking #
# with a new scoring function, efficient optimization and #
# multithreading, Journal of Computational Chemistry 31 (2010) #
# 455-461 #
# #
# DOI 10.1002/jcc.21334 #
# #
#################################################################
Using single ligand docking mode
Output will be ./keys/warfarin_out.pdbqt
Reading input ... done.
Setting up the scoring function ... done.
Search_depth is fixed to 10
Analyzing the binding site ... done.
GPU Platform: NVIDIA CUDA
GPU Device: NVIDIA GeForce RTX 3090 Ti
Using random seed: -1583172689
Build kernel 1 from source
OpenCL version: 3.0
Build kernel 2 from source
OpenCL version: 3.0
Perform docking|================done=================|
Refining ligand ./keys/warfarin results...done.
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -7.8 0.000 0.000
2 -7.5 4.709 8.310
3 -7.4 5.243 8.602
4 -7.3 0.892 2.132
5 -7.3 0.250 1.024
6 -7.2 4.941 8.109
7 -7.2 4.676 8.364
8 -7.1 2.083 4.311
9 -7.1 0.901 2.338
Writing ligand ./keys/warfarin output...done.
AutoDockVina-GPU3 total runtime = 6.150 s
Step 6: Compare results.
And there's the payoff: candidate Pocket 1 gives a much stronger ligand affinity than the pocket I guessed at on the last page (-7.8 kcal/mol vs -5.1 kcal/mol). Letting fPocket actually find the binding site instead of eyeballing it made a real difference.
Step 7: Use PyMol to view the results.
Open up PyMol, play around with the "show" settings and make it pretty. Using the terminal inside PyMol, enter the following:
load ~/Documents/MolecularDocking/locks/1H9Z.pdb, protein
load ~/Documents/MolecularDocking/keys/warfarin_out.pdbqt, targeted_dock
load ~/Documents/MolecularDocking/locks/1H9Z_out/pockets/pocket1_vert.pqr, pocket1_cloud
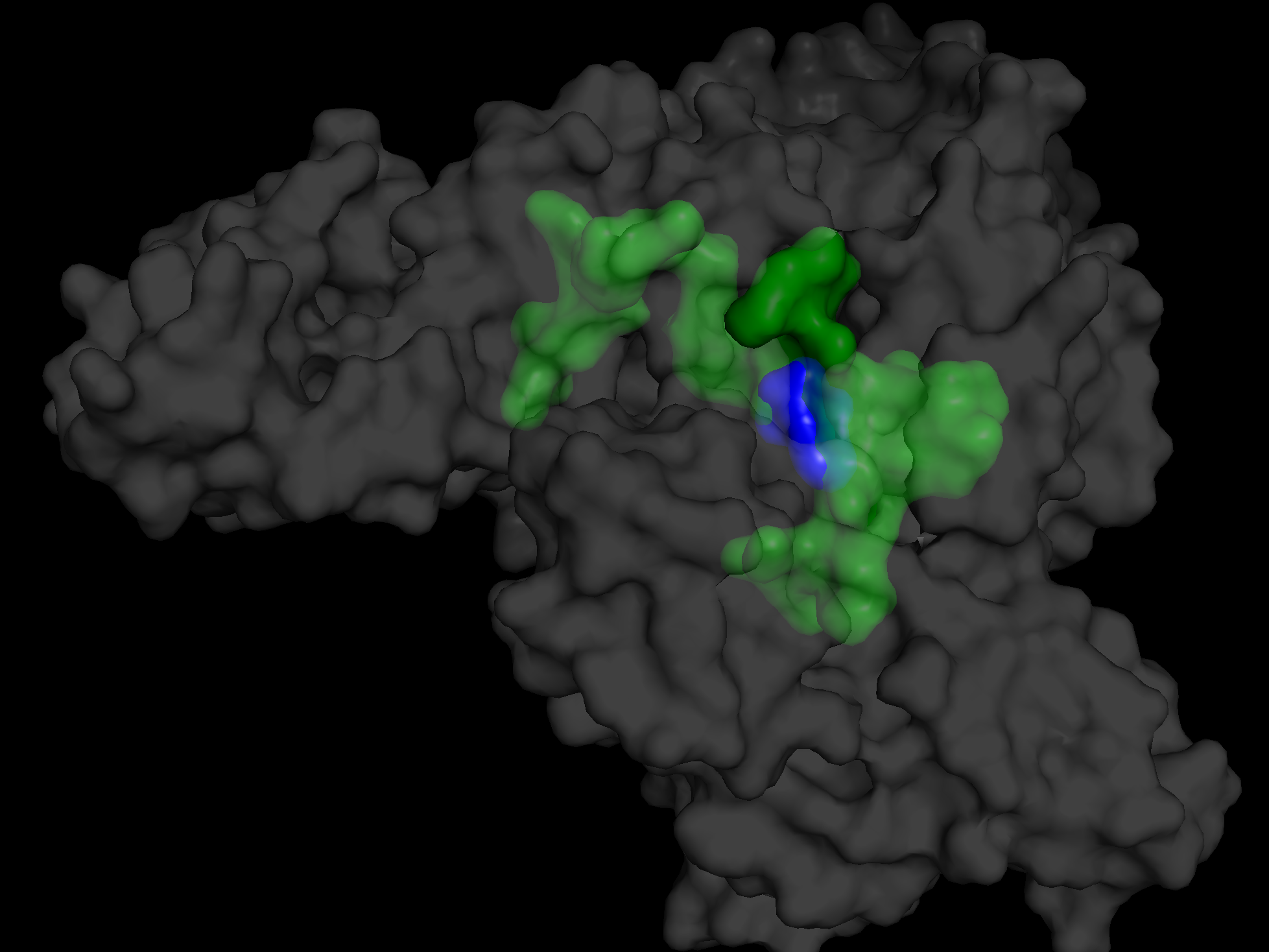 The transparent white/gray area is the protein.
The transparent white/gray area is the protein.
The transparent green area is Pocket 1.
The blue area is the ligand, warfarin.
Summary:
We have shown how to use the tools, the basic work flow, and the meaning and importance of the stats given by the tools.
- Using PyMOL to visualize molecular structures.
- Using Open Babel to convert file formats (SDF to PDBQT).
- Using AutoDock Vina-GPU to perform molecular docking.
- Using conda to manage software environments.
- Using fPocket to find candidate locations for docking.
Next we will cover more of the theory of how and why.